

Our research
Simulation methods are a powerful tool to determine the structure and electronic properties of condensed matter systems. Due to the rapidly increasing speed of computers, simulation is a very active field with many opportunities to improve simulation methods and with new applications becoming feasible. We develop better algorithms for such atomistic calculations and apply them to challenging problems. The research has interdisciplinary character, involving physics, mathematics, chemistry and computer science.
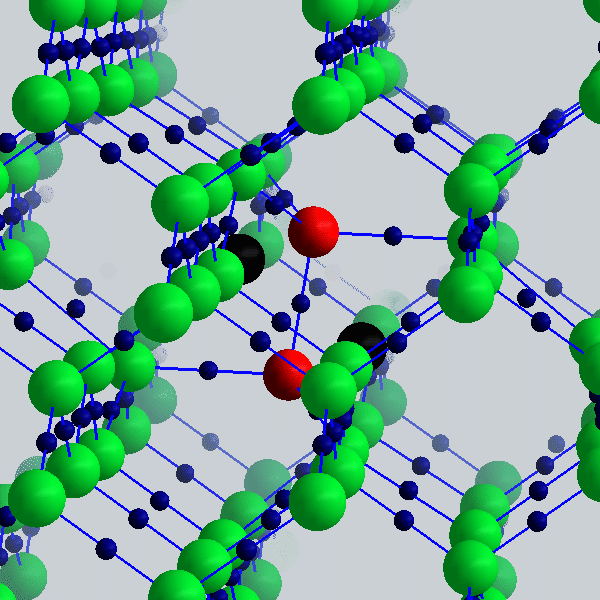
The picture on the upper right shows the lowest energy point defect in silicon. Our simulation (Goedecker et al., Phys. Rev. Lett. 88, 235501 (2002)) reveals the atomic positions (large spheres) and the bond centers (small blue spheres). Two silicon atoms that were in the crystalline bulk in the positions denoted by the black spheres moved to the positions given by the red spheres.
Finding ground state structures requires finding the global minimum of the potential energy surface. This can be done with the minima hopping algorithm (Goedecker, J. Chem. Phys 120, 9911 (2004) ). The visualization (bottom right picture) of the minima hopping algorithm shows the formation of a fullerene molecule out of a graphite sheet containing 60 carbon atoms.
A full list of publications and citations can be found on Google scholar .